- وبلاگ مرکز اوتیسم جواهرات
- اوراق قرضه ایالات متحده محافظت شده از تورم تجمع کرده است اما همه داستان را خریداری نمی کنند
- خطوط و پیش بینی های ریاضی فارکس
- الگوی صخره ای Hikkake - نحوه ضرب و شتم بازار
- 3 ستون عملکرد تجارت
- 5 راه برای رشد تجارت خود در دنیای Covid-19
- برنامه مربی و کارشناسی ارشد Okex Beacon برای ایجاد فرصت های شغلی در بخش رمزنگاری
- تورنتو آبی جیز در مدیریت بار کار هارون سانچز خلاق است و پارچ را به کلاس یک Dunedin ارسال می کند
- معاملات را بالا ببرید معاملات را بالا ببرید معاملات
- سایه طولانی در شمع تأیید برک آوت

آخرین مطالب
امکانات وب


خوشه بندی با محاسبه فاصله بین هر جفت واحد که می خواهید خوشه بندی کنید ، شروع می شود. یک ماتریس فاصله متقارن خواهد بود (زیرا فاصله بین x و y همان فاصله بین Y و X است) و صفر روی مورب خواهد داشت (زیرا هر مورد از خود فاصله صفر است). جدول زیر نمونه ای از ماتریس فاصله است. فقط مثلث پایین نشان داده شده است ، زیرا مثلث فوقانی را می توان با بازتاب پر کرد.
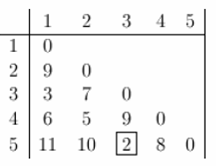
حالا اجازه می دهیم خوشه بندی را شروع کنیم. کوچکترین فاصله بین سه تا پنج است و آنها ابتدا به یک خوشه "35" پیوند می یابند.
برای به دست آوردن ماتریس فاصله جدید ، باید 3 و 5 ورودی را حذف کنیم و آن را با ورودی "35" جایگزین کنیم. از آنجا که ما از خوشه بندی ارتباط کامل استفاده می کنیم ، فاصله بین "35" و هر مورد دیگر حداکثر فاصله بین این مورد و 3 و این مورد و 5 است. به عنوان مثال ، D (1،3) = 3 و D (1، 5) = 11. بنابراین ، D (1 ، "35") = 11. این ماتریس فاصله جدید را به ما می دهد. موارد با کمترین فاصله در مرحله بعدی خوشه بندی می شوند. این 2 و 4 خواهد بود.

ادامه این روش ، پس از 6 قدم ، همه چیز خوشه بندی می شود. این در زیر خلاصه شده است. در این طرح ، محور y فاصله بین اشیاء را در زمان خوشه بندی نشان می دهد. به این ارتفاع خوشه گفته می شود. تجسم های مختلف از اقدامات مختلف ارتفاع خوشه استفاده می کنند.
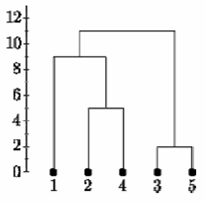
پیوند کامل
در زیر دندروگرام تک پیوند برای همان ماتریس فاصله قرار دارد. این کار با خوشه "35" شروع می شود اما فاصله بین "35" و هر مورد اکنون حداقل D (x ، 3) و D (x ، 5) است. بنابراین C (1 ، "35") = 3.
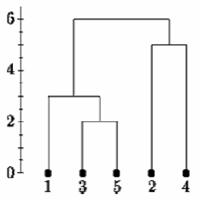
تعیین خوشه ها
یکی از مشکلات خوشه بندی سلسله مراتبی این است که هیچ روش عینی برای گفتن چند خوشه وجود ندارد.
اگر درخت پیوند واحد را در نقطه ای که در زیر نشان داده شده است ، برش دهیم ، می گوییم که دو خوشه وجود دارد.
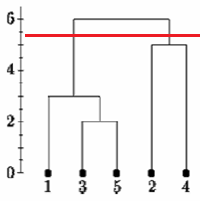
با این حال ، اگر درخت را پایین بیاوریم ممکن است بگوییم که یک خوشه و دو تک آهنگ وجود دارد.
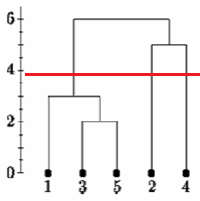
هیچ روش متداول برای تصمیم گیری در مورد کجا برای بریدن درخت وجود ندارد.
بیایید به برخی از داده های واقعی نگاه کنیم. در تکالیف 5 بیان ژن را در 4 منطقه از 3 مغز انسانی و 3 مغز در نظر می گیریم. RNA به ریزگردهای بیان ژن انسانی Affymetrix ترکیبی شد. ما داده ها را با استفاده از RMA نرمال کردیم و با استفاده از لیمما ، تجزیه و تحلیل بیان دیفرانسیل انجام دادیم. در اینجا ما 200 ژن قابل توجه متفاوت از مطالعه را انتخاب کردیم. ما تمام ژنهای متفاوت بیان شده را بر اساس میانگین بیان آنها در هر یک از 8 گونه توسط درمان های منطقه مغزی خوشه می کنیم
در اینجا خوشه های مبتنی بر فاصله اقلیدسی و فاصله همبستگی ، با استفاده از خوشه بندی کامل و تک پیوند وجود دارد.
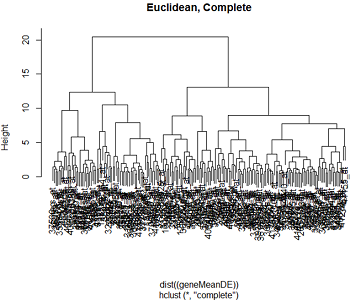
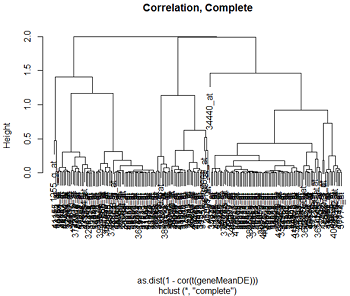
ما می توانیم ببینیم که الگوی خوشه بندی برای مسافت پیوند کامل تمایل به ایجاد خوشه های جمع و جور خوشه ها دارد ، در حالی که پیوند واحد تمایل به اضافه کردن یک نقطه به یک زمان به خوشه دارد و باعث ایجاد خوشه های بلند می شود. همانطور که ممکن است از بحث ما در مورد مسافت انتظار داشته باشیم ، فاصله اقلیدسی و فاصله همبستگی باعث ایجاد دندروگرام بسیار متفاوت می شود.
خوشه بندی سلسله مراتبی به ما نمی گوید که چه تعداد خوشه وجود دارد ، یا اینکه باید دندروگرام را برای تشکیل خوشه ها برش دهیم. در R یک Cuttree تابعی وجود دارد که یک درخت را در یک ارتفاع مشخص به خوشه ها می رساند. با این حال ، بر اساس تجسم ما ، ممکن است ترجیح دهیم شاخه های طولانی را در ارتفاعات مختلف برش دهیم. در هر صورت ، یک ذهنیت نسبتاً عادلانه در تعیین اینکه شاخه ها باید و نباید برش دهند برای تشکیل خوشه های جداگانه وجود دارد.
درک خوشه ها
برای درک خوشه ها ، معمولاً مقادیر log2 (بیان) ژن ها را در خوشه ترسیم می کنیم ، یا به عبارت دیگر ، عبارات ژن را روی نمونه ها ترسیم می کنیم.. این توطئه های پروفایل نامیده می شوند.
در اینجا برخی از توطئه های پروفایل از خوشه بندی ارتباط کامل هنگامی که از فاصله اقلیدسی استفاده کردیم:
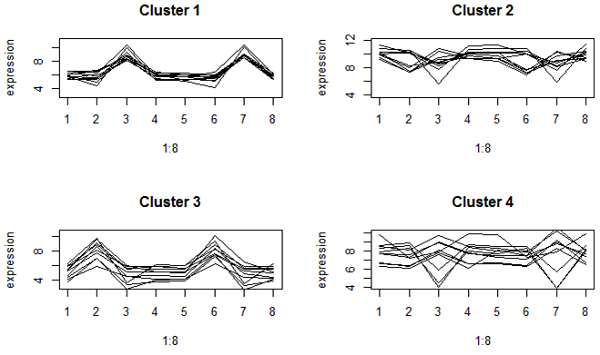
اینها بسیار محکم به نظر می رسند. با این حال ، خوشه های 2 و 4 ژن هایی با الگوهای مختلف بالا و پایین دارند ، زیرا تقریباً یک بیان متوسط دارند. خوشه 2 ژنهای بسیار بیان شده ای هستند.
این چیزی است که ما هنگام استفاده از فاصله همبستگی به دست آوردیم:
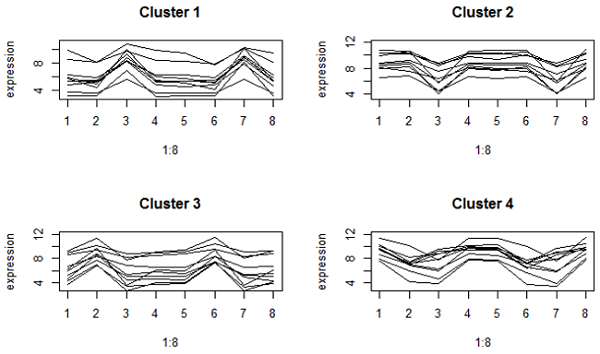
اینها در محور y بسیار سست تر هستند زیرا همبستگی بر الگوی بیان متمرکز است ، نه میانگین. با این حال ، تمام ژنهای موجود در یک خوشه در یک اوج یا دره در همان درمان ها (که مناطق مغز توسط ترکیب گونه ها هستند) دارند. خوشه های 1 و 2 ژنهایی هستند که در مقایسه با سایر مناطق مغز در هر دو گونه در مخچه بالاتر یا پایین هستند.
انتخاب لیست ژن
در اصل می توان همه ژن ها را جمع کرد ، اگرچه تجسم یک دندروگرام بزرگ ممکن است مشکل ساز باشد. معمولاً ، نوعی تجزیه و تحلیل اولیه ، مانند تجزیه و تحلیل بیان دیفرانسیل برای انتخاب ژن ها برای خوشه بندی استفاده می شود. دلایل خوبی برای انجام این کار وجود دارد ، اگرچه برخی از احتیاط ها نیز وجود دارد.
به طور معمول در بیان ژن ، متریک فاصله مورد استفاده فاصله همبستگی است. فاصله همبستگی همان محوریت و مقیاس بندی داده ها و سپس استفاده از فاصله اقلیدسی است. هنگامی که اثرات درمانی سیستماتیک وجود دارد ، ما انتظار داریم که تنوع بیان ژن از درمان تا درمان ترکیبی از اثرات درمانی سیستماتیک و نویز باشد. هنگامی که هیچ اثر درمانی وجود ندارد ، تنوع بیان ژن فقط به دلیل سر و صدا است. با این حال ، مرکزیت و مقیاس گذاری داده ها تمام متغیرها را در همان مقیاس قرار می دهد. از این رو ژن هایی که به دلیل شانس الگوی خود را نشان می دهند ، از آنهایی که دارای یک مؤلفه سیستماتیک هستند ، قابل تشخیص نیستند.
همانطور که دیدیم ، فاصله همبستگی نسبت به فاصله اقلیدسی برای مطالعات بیان ژن ، تفسیر بیولوژیکی بهتری دارد ، اما همان مقیاس بندی که باعث می شود برای یافتن الگوهای بیولوژیکی معنی دار تنظیم ژن مفید باشد ، نتایج فریبنده ای را برای ژنهایی که به طور متفاوتی بیان نمی کنند ، ارائه می دهد. انتخاب ژن ها بر اساس تجزیه و تحلیل بیان دیفرانسیل ژنهایی را که احتمالاً فقط الگوهای شانس دارند ، از بین می برد. این باید الگوهای موجود در خوشه های ژن را تقویت کند.
با این حال ، به عنوان یک احتیاط ، اثرات انتخاب ژن را بر روی نمونه ها یا درمان های خوشه بندی در نظر بگیرید. ژنهای منتخب آنهایی هستند که در تجزیه و تحلیل بیان دیفرانسیل مثبت هستند. استفاده از آن ژن ها برای نمونه های خوشه ای به سمت خوشه بندی نمونه ها با درمان مغرضانه است.
- نسخه دوستانه چاپگر

ما را در سایت فارکس کاران ایران دنبال می کنید
برچسب :
نویسنده : ديناروند فهيمه
بازدید : 31
